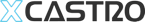
Design and development of medical products from concept to manufacture, including documentation to CE and FDA requirements as well as design verification and validation.

The medical sector is where I concentrate most of my work. The main difference in this sector is the documentation required to meet regulations — the key aspects being risk management and design verification.
My experience is mainly in CE and FDA regulations. I follow ISO 13485 and 21 CFR processes, having developed Class I, IIa, IIb and III devices with complete technical files, including design verification and human factors validation.
I am familiar with the most relevant ISO/IEC standards, including 13485 (quality), 60601 (electrical safety), 62304 (software), 10993 (biocompatibility), 14971 (risk management), and 62366 (usability).
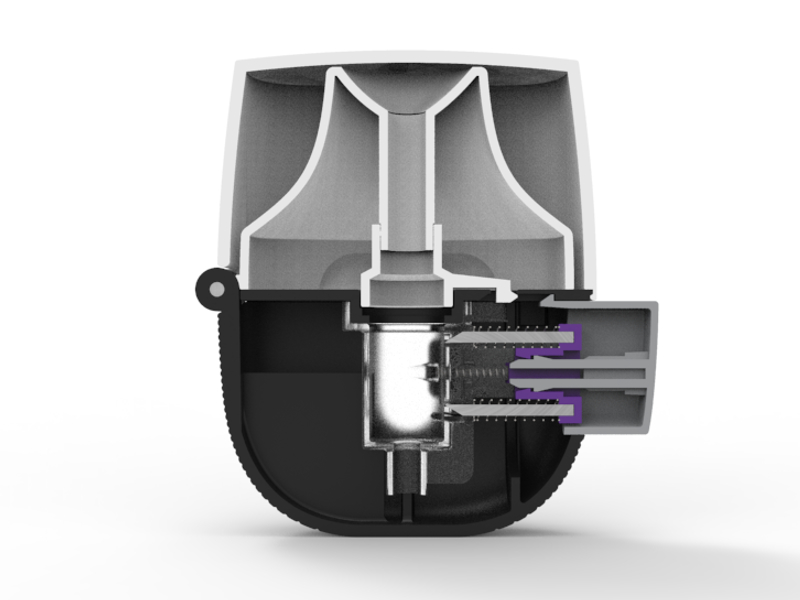
Within the medical sector, I specialize in drug delivery, wound care and medical therapy, but have also worked on diagnostics, surgical and other devices, including connected devices and electromechanical systems.
Maximum attention to detail and understanding of complex regulatory requirements.
Understanding of advanced technologies that require high-tolerance processes.
Patient outcome and experience as a priority, to deliver the ideal treatment.
Precision, ergonomics and cost that improve complex procedures.
Understanding patients' concerns and responding with appealing solutions.
Using technology wisely to deliver the best user experience and gather the required data.

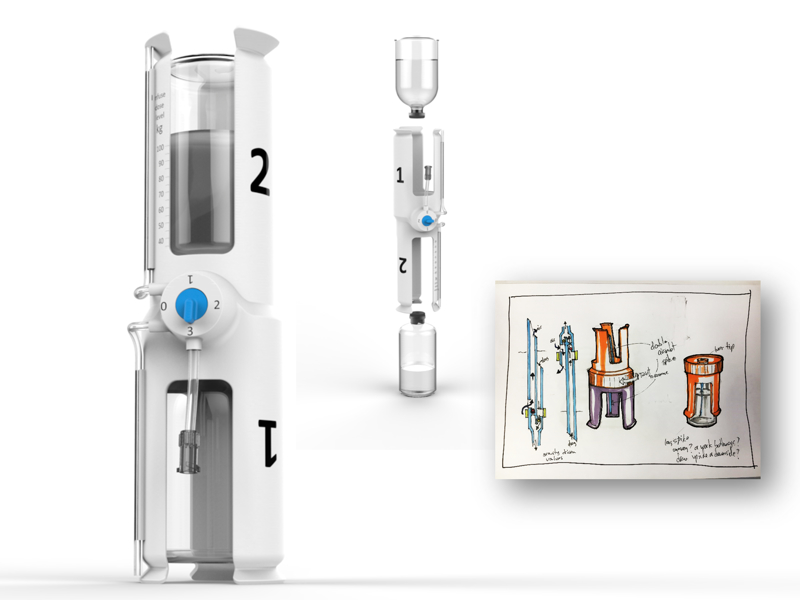
When developing medical products, it is critical to follow a rigorous documentation process to deliver a thorough technical file in compliance with regulatory requirements.
My approach follows a "V" model that aligns with ISO 13485 and 21 CFR. This model sets out documentation and quality requirements from an early stage, including a detailed look at risk management following ISO 14971, and the required verification and validation strategy from the outset.
My experience isn't limited to design and engineering — I can also assist companies and clients with their regulatory documentation, technical file, and design verification process.
Connectivity has become a key technology area that can improve patient compliance and behaviour. I have worked on the design of several connected drug delivery devices, developing different platforms and technologies to track how a device has been used and help patients learn to use it efficiently.
Connected devices require special attention in several areas: the technical side, such as using the right sensors and optimizing energy use to reduce battery size; efficient use of electronics and software for sleek performance and communication; and finally, an intuitive and elegant interface that engages patients.

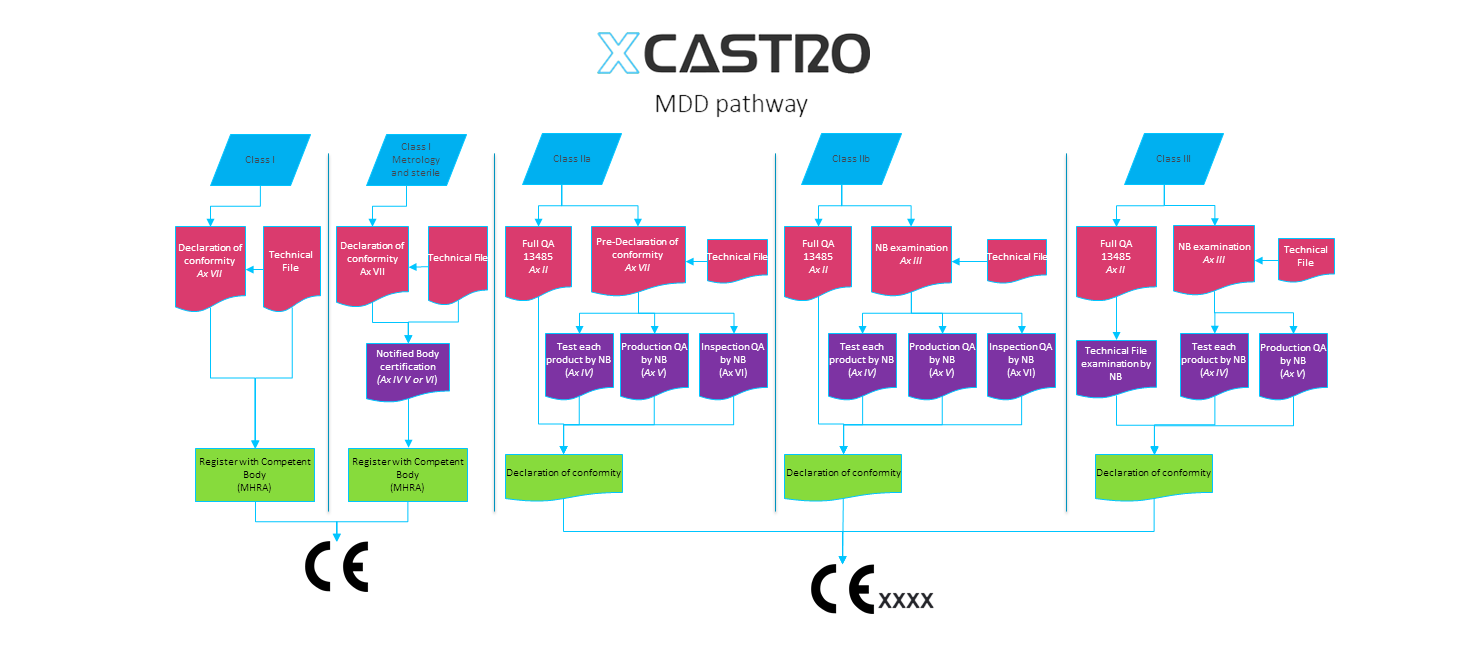
A critical aspect of medical device development is having a clear understanding of the regulatory process, documentation requirements, and a defined regulatory strategy.
The chart below illustrates how complex the regulatory documentation workflow can be, and how good planning in this area helps deliver the required documentation at each stage.
This is perhaps the most critical aspect of a medical device development programme, as it demonstrates that the product is fit for purpose and safe to use. Testing must follow a series of standards, with validated protocols using Gauge R&R or other methodologies, and calibrated equipment. Planning in this area is essential — a good verification strategy and plan considerably reduces cost and timescale. My experience in this area spans from drug delivery devices to Class III casework.

Regulations have become more demanding in requiring that devices are safe to use and free from use error. This is a relatively new field with no single fixed process to follow — each device may require a different usability strategy. I have developed human factors reports including both formative and summative studies.
In my experience, the key to human factors work is the quality of the studies, not the quantity, and conducting them at the right development phase rather than with every iteration. Formative studies should remain flexible and fast, while summative studies need to be rigorous and exhaustive.
I support organizations from start-ups to multinational pharmaceutical companies on different aspects of product development and design team leadership. I can collaborate on a contract, consultancy, or associate basis. I am based in Cambridge, UK.
Find out more on how I work, or contact me.
Tell us about the project — what stage it's at, and what you need help with.
Or email directly:
xorge@x-castro.com