")
")
Xorge Castro
Industrial Design, Mechanical Engineering, Manufacturing and Management
Design engineer consultant based in London (Islington, Camden, and Hackney) with 20 years of experience in product development as Industrial Designer, Mechanical Engineer, Usability Engineer, Human Factors Engineer, and Project Manager. Experienced in designing and developing medical devices, consumer electronics, FMCG and household products.
Also working as Mechanical Engineer contractor in London and Freelance Industrial Designer, engaging in projects from research and concept generation through manufacturing and NPI.


























Experienced working with technology start-ups, consultancies, multinationals and as independent consultant. Currently I am leading full development programs with multidisciplinary teams in the consumer electronic sector, as well as leading design and engineering activities.
My main motivation is to develop innovative and breakthrough products that make our lives better. I always look into understanding the user and consumer needs to develop the best technical solution for each problem, without ever losing the vision for commercial success.
I was trained as Industrial Designer and gain an MPhil in Industrial Systems by the University of Cambridge. My experience in engineering has been through working in different sectors and developing different technologies suing a wide range of materials and processes. As a Designer, I have been involved in all the design process, from uncovering user needs and commercial opportunities, throughout developing consumer products to the slightest detail.
In this website I showcase some of my experience and capabilities, hope you find it interesting and please get in touch if you have any comments. I work with clients across the world, but mainly in London but mainly in London; in Hackney, Islington and Camden and Cambridge.
















Further work examples
I always follow a stage gate/ phased that allows me to identify any design risks early on and solve them to develop products in the required timescales, budget and quality.

Areas of Expertise
I work across many fields in the development of new products and technologies; from identifying user needs all through setting a production line. In my career I have lead and managed projects that required special attention in each of these fields and have developed sufficient experience and knowledge in each of them in order to be able to either conduct projects on my own or oversee teams.
Industrial Design
Create the ideal style and aesthetic solution that will align with a brand identity and appeal to the consumer’s desires.

Mechanical Engineering
Produce elegant, efficient and innovative technical solutions that will enhance the user experience.
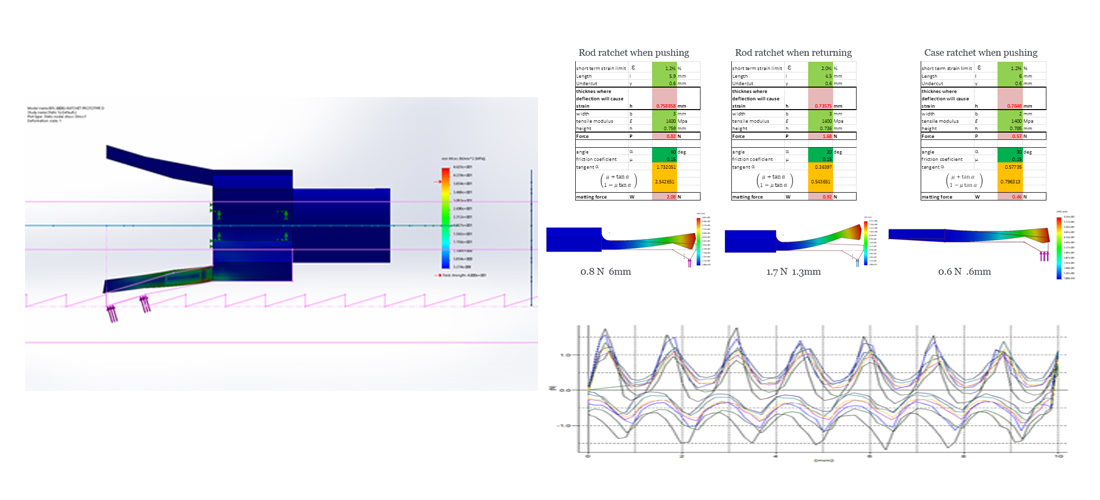
Manufacturing
Identify the best materials, processes and suppliers that will meet the tightest requirements in an ever evolving sector.

Usability
Design products that are easy, safe, intuitive and natural to use.

Medical Devices
Work in compliance with FDA and CE regulations to develop new healthcare products.
.jpg)
Consumer insight
Identify unmet needs and how products can improve user’s experiences.
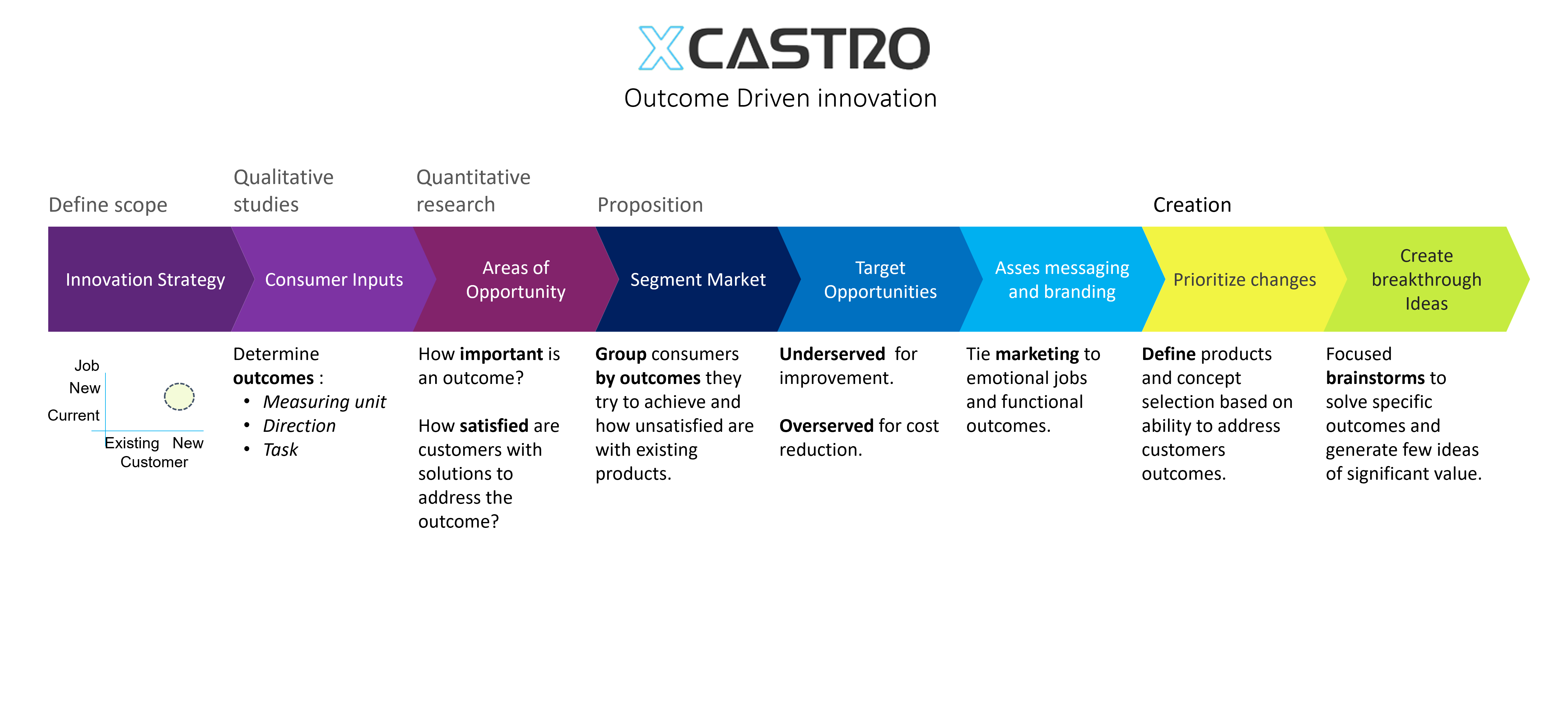
Design Process
All design and engineering development work should follow a process in order to manage risks and meet deliverables. This process starts by stablishing clear objectives and dividing the work into manageable phases. This will then allow to plan a whole project and each stage.
In my career I have worked across all phases of the development and used different approaches. Through this journey I have adapted and developed a Stage-gate development process that aligns with most companies. Each phase looks into de-risking specific aspects of the product or technology to ensure a successful outcome.
These process can comply with FDA, 13485 and CE regulations for medical products, as well as aligning with NASA’s Technology readiness Levels (TRL). The secret, is to achieve a level of confidence at each stage that the product will work, so that all the critical work is conducted at the early stages and the main risks are controlled as investments increase in the last phases.
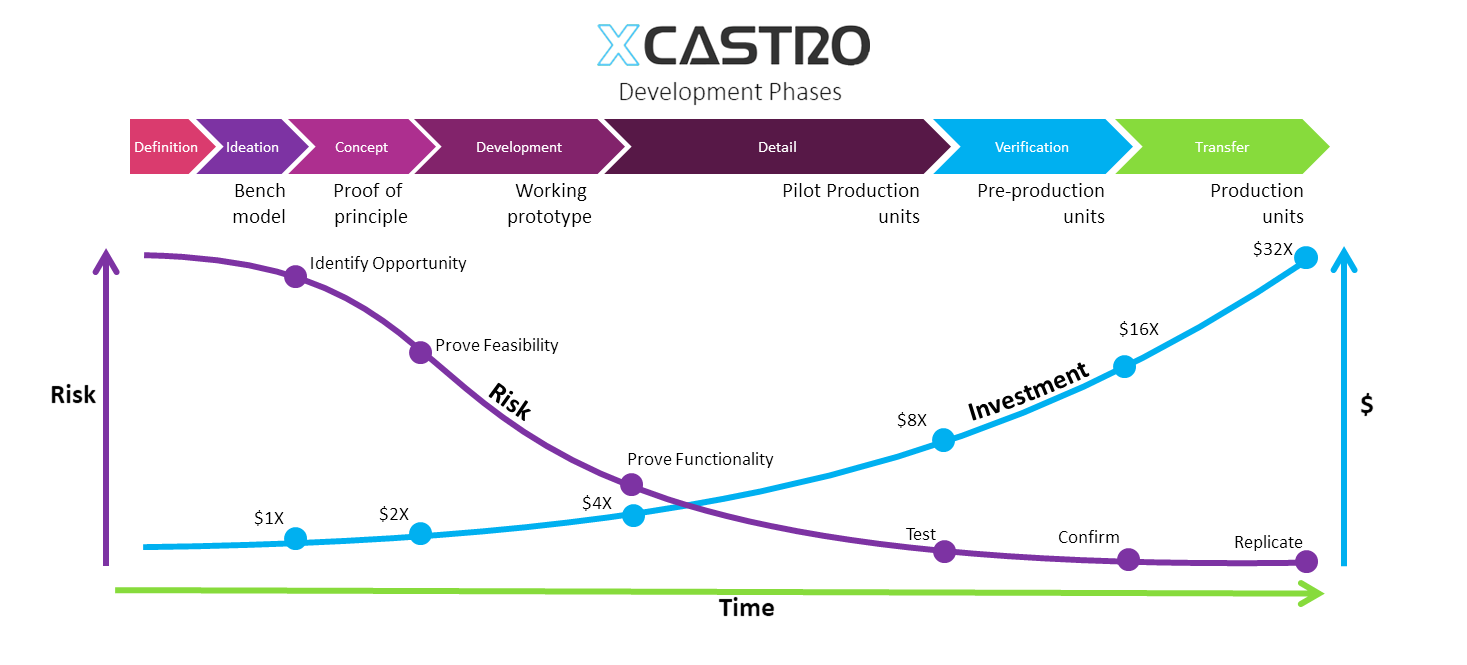
Product Development
Developing a new product requires a good understanding on design and engineering. My experience in both fields allows me to deliver high quality results in tight timescales and within a set budget. I believe that a good product development plan requires a perfect balance between all disciplines involved. With the challenge that all programmes are unique and demand different efforts at each stage. The key to great design is to understand this balance and pay enough attention at each required challenge at the right time.
A great products has to be useful, initiative, beautiful and accessible.
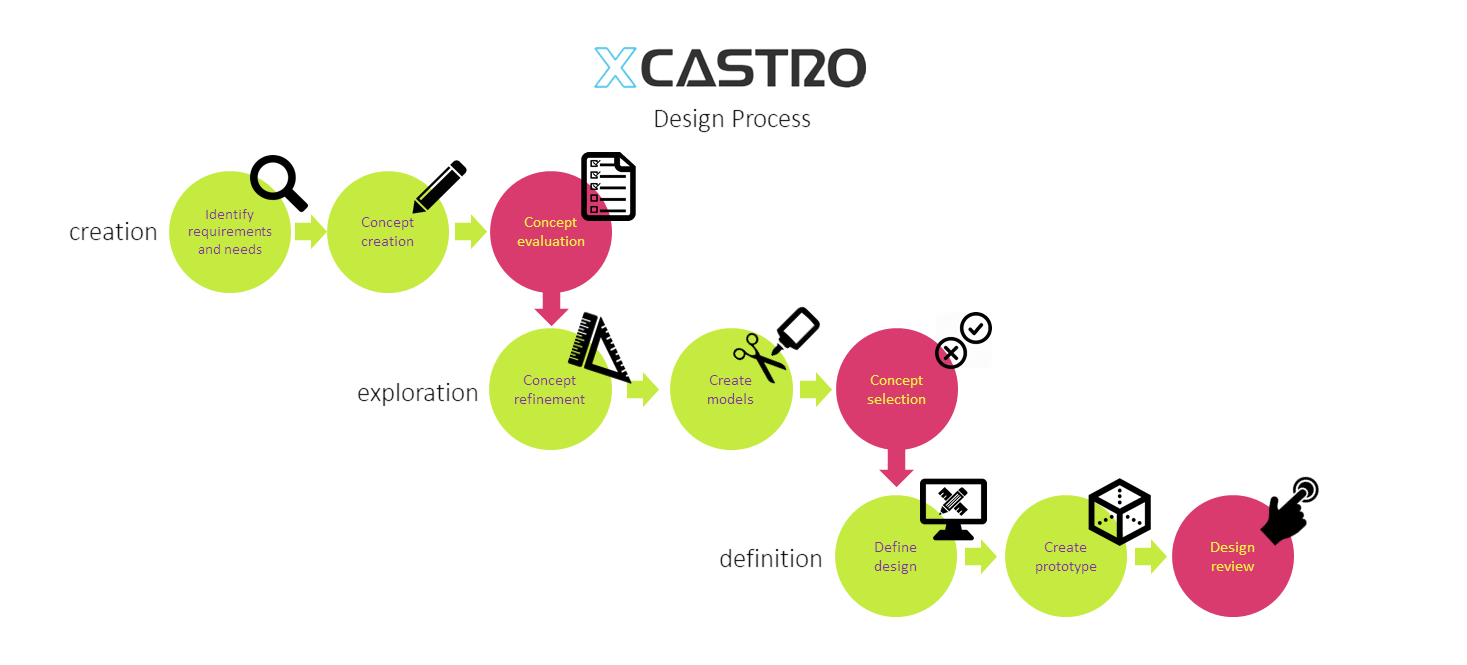
Innovation
In my day to day work I need to be able to uncover opportunities for innovation and foresee technical challenges. To create a great products, it is paramount to understand the user and the commercial landscape and be able to translate these observations into innovation opportunities. On the other hand, it is critical to understand technical limitations and boundaries, to known when and how to push them to the edge in order to deliver breakthrough innovation.

Design process
Entrepreneurs often underestimate the required level of risk and challenges that can be faced in the journey of designing a new product. The main one, is that it is presumed that once a concept has been developed, then the rest of the work is straightforward, simple and cheap.
The reality is that if detailing, engineering, manufacturing and testing is not conducted correctly, it could cause severe delays, substantial expenses and in many cases the venture to fail.
My understanding and experience across all the design journey allows me to clearly plan a whole development programme that will align to individual objectives, timescales and technical challenges.

Project management and team leadership
Leading design and engineering teams requires more than just a gant chart and documenting design reviews. In my career I have seen may perfectly managed projects with the best plans and record keeping that delivered products that failed commercially or never made it to market.
What these projects lacked, was a good technical understanding of the mechanical and manufacturing challenges and great vision and knowledge of the commercial requirements. In other words, the ability to adapt and lead as soon as risks are uncovered. As well as knowing where to find this risks.
Most of my latest work has been on exactly in this field, sometimes defined as System engineer, project manager, lead designer or team leader. My track record of delivering successful programmes of work demonstrates that my approach in this field will guarantee an effective outcome in any new product development project.

About me
My name is Xorge Castro, I work as a product design engineer, Mechanical Engineer and Project Manager in Cambridge and London in Islington, Hackney and Camden. My focus is on creating innovative products that improve our lives. I work as contract, freelance and consultant for the medical and consumer sector. My experience combines an industrial design formation with a deep experience in mechanical engineering.
Find out more on how I work or send me an email